心血管疾患の予防と治療における新たな洞察
かつてないスピードと影響力で、遺伝子治療の黄金時代がやってきます!近年、さまざまな遺伝子治療薬が世界的に承認され、その多くが臨床試験段階にあります。遺伝子治療は、多くの患者に新しい希望を与えています。
遺伝子治療薬の創製は、十分な基礎研究の上に成り立っています。革新的で完成度の高い基礎研究は、遺伝子治療の臨床研究にも活かされる可能性があります。本日は、当社のクライアントが発表された研究論文を例に、遺伝子治療の前臨床研究の考え方をご紹介します。

研究背景
心肥大は心血管系疾患の初期症状であり、その後、拡張機能障害や不整脈を引き起こし、最終的には心不全に至る可能性があります。したがって、病的な心肥大の主要な分子メカニズムを理解することは、新しい治療標的や治療戦略を開発するために極めて重要です。
ハルビン医科大学付属がん病院の研究者らは、III型トランスフォーミング増殖因子β受容体(TβRIII)が心肥大の病態形成に重要な制御的役割を担っていることを発見しました。TβRIIIは、β-アレスチン2依存性CaMKIIの活性化を介して、心肥大を誘発します。研究者らは、AAV9ベクターを介した特異的shRNAを用いてTβRIIIの発現を低下させたところ、心肥大に伴う症状が効果的に緩和されることを見出しました。この研究により、心肥大を制御するための新たな治療標的および戦略の開発につながることが期待されます。
研究構想
1. 心肥大組織におけるTβRIIIの異常発現を発見。
2. TβRIII過剰発現マウスを構築し、TβRIIIの高発現がマウスの心肥大を誘発することを発見。
3. AAV9-shTβRIII を注射して TβRIII の発現を低下させた後、イソプロテレノール(ISO)注入または横大動脈狭窄(TAC)により心肥大を誘発。TβRIIIの発現低下により、心肥大や心機能障害などの症状が緩和されることを発見。
4. メカニズム解明:TβRIIIはβ-アレスチン2によるCaMKIIの活性化を介して心肥大を誘発することを発見。
研究結果
1.ヒトおよびマウス心肥大検体におけるTβRIIIの発現について
著者らはまず、ヒト心肥大検体におけるTβRIIIの発現プロファイルを検討しました。その結果、心房性ナトリウム利尿ペプチド、脳性ナトリウム利尿ペプチド、β-MHCなどのバイオマーカーと同様に、ヒト心肥大検体においてTβRIIIの有意な発現上昇が認められました(図1A、1B、1C)。次に、マウスにISO注入またはTACにより心肥大を誘発し、左心室(LV)におけるTβRIIIの発現が有意に増加することを見出しました(図1Dおよび1E)。さらに、免疫蛍光染色により、TβRIIIがマウスLV組織で発現していることが明らかになりました(図1F、a)。蛍光強度のラインスキャン分析により、TβRIIIは主に成熟心筋細胞の細胞膜近傍に存在することが示されました(図1F、b)。これらの結果から、TβRIIIの発現増加は、心肥大の発生に関与している可能性が示唆されました。
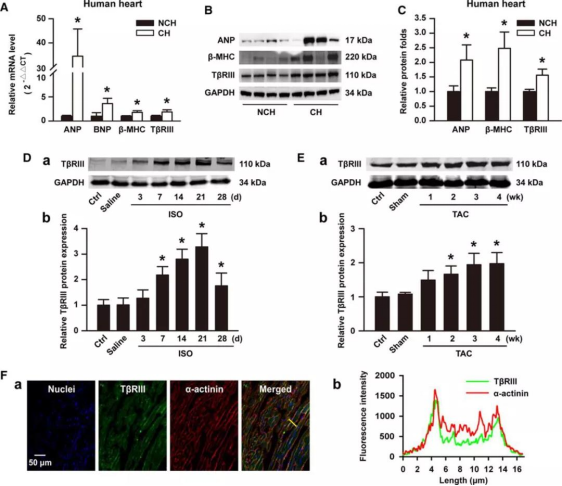
図1. 心肥大検体におけるTβRIIIの発現増加
2.TβRIII過剰発現によるマウスの心肥大の誘発
研究者らは、心臓特異的TβRIIIトランスジェニック(CS-TβRIII-Tg)マウスを作製し、動物モデルで心肥大などの表現型を再現しました。心臓特異的α-MHCプロモーターを用いてマウスの心臓にTβRIIIを過剰発現させ(図2A)、ウェスタンブロッティング(WB)と定量ポリメラーゼ連鎖反応(qPCR)(図2B、2C)により、マウスモデルの確立に成功しました(サイヤジェン社によって確立された)。その後、CS-TβRIII-Tgマウスモデルを用いて一連の実験が行われました。
野生型(WT)マウスと比較して、心房性ナトリウム利尿ペプチド、脳性ナトリウム利尿ペプチドおよびβ-MHCタンパク質のレベルは、それぞれCS-TβRIII-Tgマウスにおいて増加しました(図2D)。CS-TβRIII-Tgマウスでは、連続波エコー図を用いて、LV壁の厚さおよびLV質量の増加が検出されました(図2E)。組織学、心臓重量/体重の比、および心臓重量/脛骨長の比較分析により、TβRIIIの心臓特異的過剰発現は、自然発生的な心肥大を引き起こすことが示されました(図2F、図2G、および図2H)。また、ISOやTACで処理した場合、CS-TβRIII-Tgマウスの心肥大の程度は野生型マウスよりも深刻でした(ISO:図2I~2K;TAC:図2L~2N)。以上のことから、TβRIIIの過剰発現は心肥大の誘発に十分であり、TβRIIIの発現上昇はストレス誘発性心機能障害を促進することが明らかとなりました。
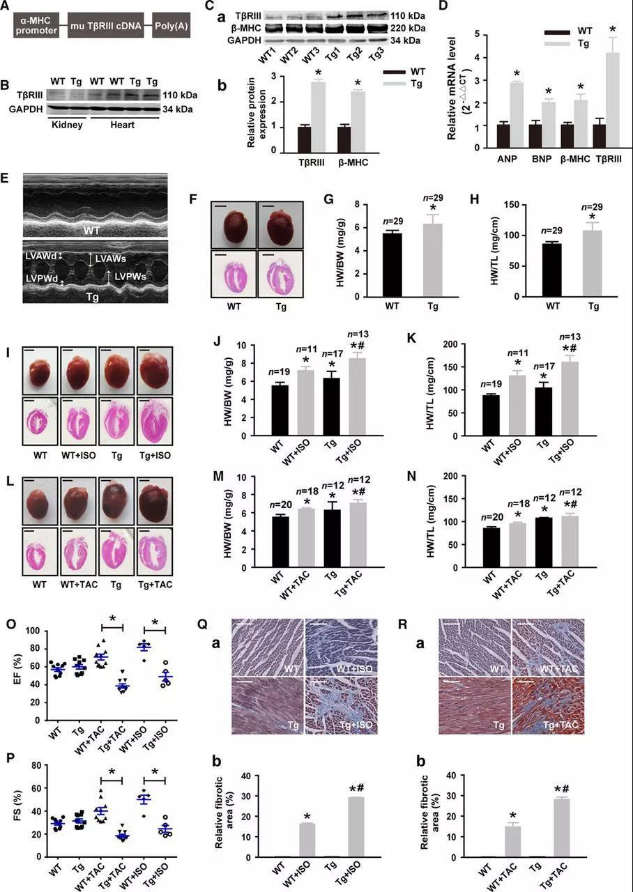
図2.TβRIIIの過剰発現による心肥大の促進
3.TβRIIIの発現抑制によるストレス誘発性心肥大症状の緩和
著者らは、TβRIIIの過剰発現は心肥大を促進することから、TβRIIIの発現を低下させることでストレスによる心肥大を軽減できるのではないか、と推測しています。そこで、心臓を標的としたAAV9ベクターによる特異的shRNA(AAV9-shTβRIII)を用いて、マウス心臓のTβRIII遺伝子をノックダウンし、WBによりその表現型を検証しました(図3B)。
WTマウスと比較して、shTβRIIIマウスではISOまたはTAC処理により誘発される肥大指数が有意に減少し、心肥大および心機能障害が緩和されました。(図3C)。また、LV質量/体重、LV質量/脛骨長の比を算出することにより、TβRIIIのノックダウンが心肥大指数を低下させることも示されました(図3D〜図3G)。さらに、TAC手術の12週間後、WTおよびAAV9を介したスクランブルshRNA感染マウスの両方で、駆出率(EF)および左室内径短縮率(FS)が有意に低下し、これはTβRIIIの発現を低減することにより、TACによる心筋収縮機能障害を緩和することを示唆しています(図3Hおよび図3I)。また、TβRIIIの心臓特異的ノックダウンにより、ISOまたはTACで誘発される心肥大マーカーβ-MHCタンパク質および心房性ナトリウム利尿ペプチド、脳性ナトリウム利尿ペプチド、β-MHC mRNAの発現量が有意に減少しました(図3J~図3L)。
本研究では、研究者らは、AAV9ベクターを介した特異的shRNAを用いて、マウスにおける内因性TβRIIIの発現を低下させ、shTβRIIIマウスでは対照マウスと比較して、心肥大症の表現型を有意に低下させることを確認しました。これは、TβRIIIの発現量を低下させることで心機能障害を改善できることを示すとともに、心肥大の発症機序におけるTβRIII機能を検証するものでもあります。
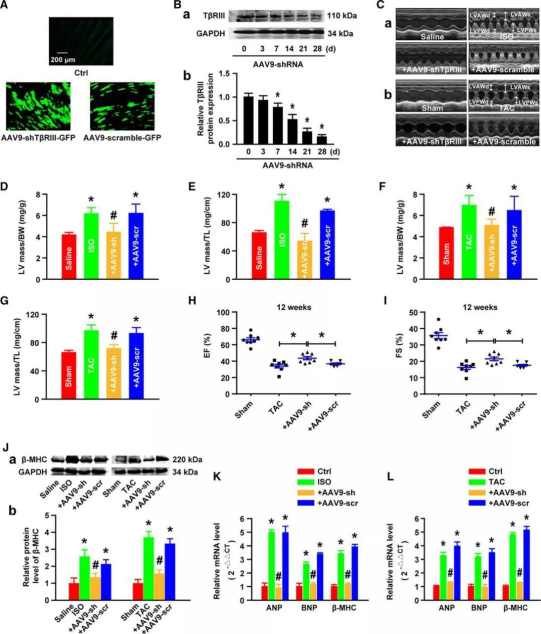
図3.TβRIII発現低下による心肥大の緩和
4.TβRIIIはβ-アレスチン2によるCaMKIIの活性化を介して心肥大を誘発
解析の結果、TβRIIIはβ-アレスチン2と相互作用し、様々ながん細胞において増殖や遊走に影響を与えること、また、イソプロテレノールがカルモジュリン依存性タンパク質キナーゼII(CaMKII)の活性化において重要な役割を果たすことが判明されました。そこで、著者らは、in vivo および in vitro の研究を行い、最終的に TβRIII が β-Arrestin2 を介して CaMKII と相互作用し、心肥大を誘発することを明らかにしました。
結論
本研究では、著者らによって、TβRIII がβ-アレスチン2依存性CaMKIIの活性化を介して心肥大を誘発することを示す、実質的かつ正確なデータが提供されました。この発見は、心肥大のメカニズムのさらなる理解に寄与し、AAVデリバリーシステムによる心筋細胞におけるTβRIIIの発現阻害が、心肥大の予防と治療につながる可能性をも提示しました。
研究を加速させるサイヤジェン遺伝子治療プラットフォーム
サイヤジェンは、包括的なソリューションプロバイダーとして、革新的なCROプラットフォームを確立し、研究者に最先端の遺伝子組換えモデルサービスを提供しています。遺伝子治療開発のための「ワンストップソリューション」を提供し、あらゆるレベルの研究ニーズに対応します。CRISPR Cas9スクリーニング&ターゲットバリデーション、カスタムマウスモデル作製、アデノ随伴ウイルス(AAV)、レンチウイルス(LV)、アデノウイルス(ADV)の効率的なアデノウイルス/レンチウイルスのパッケージング、薬理・薬力学(PD)研究、など様々なサービスを展開しています。私たちのサービスが、遺伝子治療研究成果の効率化につながることを願っています。

参考文献:
Lou, Jie, et al. “Type III Transforming Growth Factor-β Receptor Drives Cardiac Hypertrophy Through β-Arrestin2-Dependent Activation of Calmodulin-Dependent Protein Kinase II.” Hypertension, vol. 68, no. 3, 2016, pp. 654–666.
