ハンチントン病(HD)動物モデルへの新たな洞察
ハンチントン病(HD)は、ハンチントン舞踏病とも呼ばれ、主に遺伝性の神経変性疾患です。前回、ハンチントン病について、その症状や病態変化、治療法などを紹介しました。今回は、HDの原因遺伝子、その他の治療法、関連する動物モデルの応用などを紹介し、HDの謎をさらに解き明かしていきます。
HD原因遺伝子
ハンチントン病(HD)は、ハンチンチン遺伝子(HTT)の変異型により、CAGリピートが過剰(36個以上)になり、不安定なタンパク質が形成されることで発症します。通常、CAGリピートの数は7~35個です。CAGリピート数が36~40の人は、ハンチントン病の徴候や症状を発症する場合としない場合があり、40以上の人は、普通の生活で病気を発症することになります。注目すべきは、HDの発症時期が、CAGリピート数と相関しており、リピート数が多いほど発症が早くなるということです。運動障害は通常、CAGリピート数が40個程度の人では40歳から、60個以上の人では思春期から始まります。
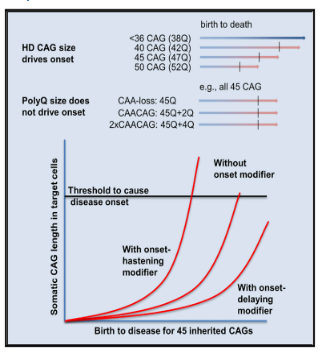
図1. CAGリピート数によるHD発症年齢の決定
HDの遺伝レベルでの原因因子は確認されていますが、その病態形成はまだ十分かつ明確に報告されていません。より一般的な説では、変異型HTTタンパク質が脳の線条体に凝集・蓄積し、線条体の神経細胞を損傷して、神経細胞のアポトーシスと線条体の萎縮を引き起こすとされています。HTT遺伝子に変異が生じると、変異型HTTの発現により、カハール小体特異的RNA(scaRNA)から形成されるRNAヘアピン、異常な反復関連非ATG(RAN)翻訳タンパク質産物、異常スプライシングにより形成されるN末端HTT遺伝子エキソン1タンパク質断片、さらにプロセシングにより生成される変異型HTTの小さな断片(mHTT)など、様々な毒性バリアントが生成されます。これらの生成物は、単量体のままであったり、オリゴマーや封入体を形成したりして、程度の差こそあれ、HD発症の一因となる可能性があります。
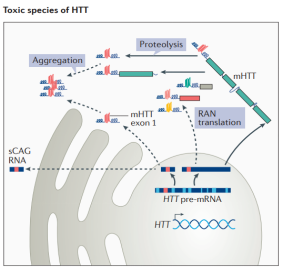
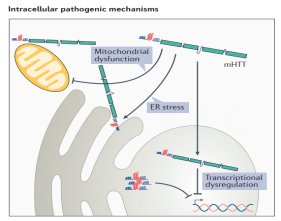
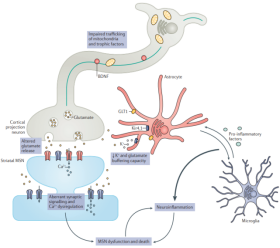
図2. HDを引き起こす複数のHTT毒性バリアント
HDでは、mHTTの存在により、転写調節障害、ミトコンドリア機能の低下、シナプス機能障害、小胞体ストレス、中型有棘神経細胞(MSN)の栄養サポートの喪失が惹起されます。mHTTは、内向き整流カリウムチャネルkir4.1およびグルタミン酸トランスポーター1(GLT1)の発現を変化させることにより、細胞外イオンの恒常性を損ない、シナプスにおけるアストロサイトによるグルタミン酸の取り込みを抑制し、ニューロンの興奮性や活性化を増加させることができます。さらに、mHTTは、末梢神経系や中枢神経系細胞の異常な免疫活性化を引き起こし、神経炎症、ひいては神経変性や神経細胞のアポトーシスを引き起こす可能性もあります。
研究治療
遺伝子治療
近年、HDの遺伝子治療は、主要な研究機関や製薬会社にとって重要な研究方向となっています。遺伝子治療には、AAVベクターによる導入、ASO薬、RNA干渉(RNAi)など、さまざまな方法があります。
現在、臨床段階にある遺伝子治療薬開発プロジェクトの一部をご紹介します。
- uniQure社が開発したAMT-130は、欧州で第1b/2相臨床試験が進行中です。AMT-130は、中枢神経系を標的とした同社初の遺伝子治療製品で、標的マイクロRNAをAAV5ベクターで送達し、mHTTの発現を大幅に低下させることができます。AMT-130は、全身麻酔下で脳の2つの特定部位(尾状核と線条体)に注入されます。これは、頭蓋骨に2〜6個の小さな穴を開け、マイクロカテーテルを通してAMT-130を投与することによって行われます。なお、侵襲的な治療法であり、一定のリスクがあることに留意する必要があります。
- ロシュ社とウェーブ・ライフ・サイエンス社の2社が開発したASO薬tominersen(トミネルセン)は、第I/II相臨床試験で重篤な副作用なく脳脊髄液中のmHTT値を有意に低下させることが示されました。しかし、第III相試験では高い有効性を示すことができず、2021年3月に第III相試験の中止が発表され、その後、第I/II相試験の他の2剤も中止となりました。
- ノバルティス社が開発した血液脳関門を通過可能な経口低分子RNAスプライシングレギュレーター・Branaplam(ブラナプラム)は、FDAからファストトラック認定を受け、現在第2相臨床試験中です。ブラナプラムは、mHTTを発現するmRNA前駆体に結合することにより、mRNAのスプライシングパターンを変化させ、mRNAの分解を誘発することができます。研究により、ブラナプラムは、in vitroの細胞モデル、動物モデルおよび初期の臨床試験において、mHTTレベルを低下させることが示されています。また、研究者らは論文の中で、この低分子療法は、RNAiやASOに加え、HD疾患進行に介入する新たな戦略であると述べています。
その他の治療法
復旦大学のBoxun Lu教授のチームは、オートファゴソーム係留化合物(ATTEC)を用いて病原性タンパク質を分解し、病気を治療するという革新的な創薬の方法を開発しました。この「低分子糊」(small molecule glue)は、HDの原因タンパク質であるmHTTに特異的に結合し、オートファジーを利用して、野生型HTTのレベルに影響を与えることなく、mHTTのレベルを低下させることができます。研究の結果、この薬剤は血液脳関門を通過することができ、少量の腹腔内注射でHDマウスの大脳皮質と線条体のmHTTレベルを著しく低下させることが示されました。この薬剤は、野生型HTTレベルに影響を与えることなく、疾患関連の表現型を改善することができ、さらに、HDの経口または注射剤開発のための入り口をも提供し、将来創薬に新たな方向性と可能性を提示するものでもあります。
動物モデル
HDマウスモデル
HDマウスモデルは、主に2つに分類されます。
一つは、トランスジェニック技術により、mHTTのN末端断片を(マウスに)過剰発現させるもので、変異HTTエクソン1の対応するタンパク質断片を発現するR6/2モデル、R6/1モデル、さらに長い断片を発現するN171-82Qモデルなどがあります。これらのN末端断片は、いずれもmHTTのポリQ配列を含んでおり、得られたN末端断片は強い毒性を持ち、凝集しやすいことも判明されています。この種のモデルは、変異遺伝子の5末端配列のみを持ち、しばしば複数コピーを持つため、HD患者とは遺伝的に矛盾します。また、表現型はしばしば数週間(数週間から十数週間)早く現れ、これも多くの患者とは矛盾します。もう一つは、完全長mHTTを、2つの方法で発現させるものです。一つは、人工染色体アプローチによるトランスジェニックヒト由来mHTTの発現であり、これには主にYAC(酵母人工染色体)とBAC(細菌人工染色体)の二つのモデルがあります。この方法の利点は、ヒ由来遺伝子が発現し、プロモーターやイントロンなどのゲノム配列など、ヒトゲノムの遺伝子の関連要素をすべて含んでいることにあります。欠点は、トランスジェニックモデルであるため、変異遺伝子のコピー数は増えても、野生型のコピー数は減らず、患者の遺伝子型と矛盾してしまう点です。もう一つは、マウスHtt遺伝子のエクソン1を患者のHTT遺伝子のエクソン1に置換して、mHTTを発現させるというものです。CAGリピード数によって、一般的に使用されるマウスモデルにはQ140、Q150、Q175モデルがあります。これらのモデルは、マウスの内因性Httプロモーターを用いてmHTTタンパク質を発現させ、本来の野生型と置き換えるという遺伝子ノックイン技術によって構築されており、遺伝学的に最もHD患者に近いとされていますしかし、この方法にも欠陥があります。エクソン1と一部のイントロン1を除き、発現している変異遺伝子はすべてマウス相同遺伝子Httであり、種差が存在エクソン一般的に、HD研究におけるマウスモデルの利点は、包括的な研究ツールと、遺伝子型および表現型が患者のものと高度に一致していること、HD病態形成や治療の研究に重要な情報提供が可能であることが挙げられます。
HD大型動物モデル
科学者たちは、HDを研究するための一連の大型動物モデルも確立しています。 その1つがHDサルモデルで、84個のCAGリピート数を持つ遺伝子エクソン1を、レンチウイルスで卵細胞に注入発現させています。ヤーキース国立霊長類研究センターのAnthony Chanと共同研究を行ったXiaojiang Li教授とShihua Li教授は、2008年にHDアカゲザルモデルを確立し、ジストニアやコレアなどの表現型の発現に成功したと報告しています。2つ目はHDヒツジモデルで、73個のCAGリピート数を持つHTT完全長cDNAを、マイクロインジェクション法で受精卵に注入しています。この方法で構築したHDヒツジモデルでは、分子レベルでいくつかのHD病理学的変化と代謝表現型を示したが、神経系機能の面では明らかな疾患表現型は観察されませんでした。3つ目は、非常に有望なHDブタモデルです。
なぜブタモデルが有望なのか?ブタの体格、系統、代謝はヒトと非常に似ているため、脳の大きさも比較的近く、ヒトと同じ溝があるからです。したがって、ブタの脳疾患モデルには独自の利点があり、臨床試験への応用が容易なのです。初期のHDブタモデルは、mHTTのN末端断片のトランスジェニック過剰発現によって構築されました。しかし、外因性の病原性遺伝子断片を発現させると毒性が強いため、生存期間が短く、病原性遺伝子を継代することができませんでした。そこで研究グループは、より正確に神経変性疾患を再現するため、CRISPR/Cas9遺伝子編集技術を用いて、ヒト変異型HTT遺伝子をブタの内在性遺伝子に挿入し、体細胞核移植によりHDノックインブタモデルを確立しました。外因性遺伝子を導入せず、内因性の野生型遺伝子を直接疾患型に変化させるため、HDの遺伝子型は患者と完全に一致しました。さらに研究を進めると、このモデルは、HD患者の線条体の中型有棘神経細胞が選択的に死滅するという典型的な病態をよく模擬でき、体重減少や異常行動といったHDに似た表現型も示されました。さらに重要なことは、これらの表現型が子孫に受け継がれることで、HD治療薬開発のための大動物モデルの安定した供給源となることです。
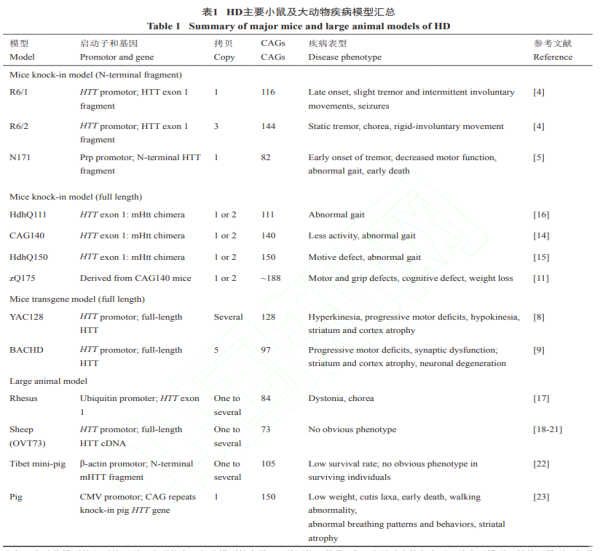
図3. HDにおける主な動物モデルの一覧
動物モデルには、上記の他に、酵母モデル、線虫モデル、ショウジョウバエモデル、ゼブラフィッシュモデルなどがあります。これらのモデルには、表現型検出のためのハイスループット実験、納期が短い、ハイスループットスクリーニングが容易であるなどの特有の利点もあります。
RDDCによる希少疾病研究へのサポート

ぜひ、公式サイト(rddc.tsinghua-gd.org)にログインしていただき、詳細をご覧ください。
*免責声明:RDDCのデータおよびツールは、科学的研究のための参考資料であり、医学的な診断や評価の結論として使用することはできません。
参考文献:
[1] https://en.wikipedia.org/wiki/Huntingtin#Gene.
[2] Zhaoyang L, Cen W, Ziying W, et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature. 2019 Nov;575(7781):203-209.
[3] Ping A, Boxun L. Current Research Status of Huntington's Disease. Chinese Journal of Cell Biology. 2018, 40(10): 1621–1632
