ハンチントン病——呪われし「舞踏」
発見と疫学
ハンチントン病(HD)は、ハンチントン舞踏病とも呼ばれ、主に遺伝性の神経変性疾患です。この病気に関する最も古い記述は、1841年にアメリカの医師であるCharles Oscar Waters(チャールズ・オスカー・ウォーターズ)によってなされたものです。その後、1872年にアメリカの医師George Huntington(ジョージ・ハンティントン)により、さらに詳しく説明されました。HDは、常染色体優性遺伝による基底核および大脳皮質の変性疾患で、臨床的には、弛緩性発症、舞踏病、認知障害、精神・行動障害を特徴とします。病気が進行すると、協調性のない不随意運動が顕著になります。身体能力は徐々に低下し、協調運動が困難となり、やがて会話もできなくなります。精神面では、通常、認知症へと移行していきます。
発症年齢は小児期から79歳までと幅広く、通常は30~50歳で発症します。20歳以前に診断されたものは若年性HDと呼ばれますが、HD全体の10%未満に過ぎません。症状は通常、舞踏運動から始まり、病気が進行するにつれて、話す、動く、考える、飲み込むなどの能力が徐々に失われていきます。10~20年程度で死に至ります。HDの有病率は105人あたり2.7人です。アジアよりもヨーロッパ、北米、オーストラリアで発症率が高いとされ、日本や台湾は欧米の1/10程度ですが、中国では3万〜4万人とされています。
病理学的変化
研究員によれば、HD患者では、線条体、大脳皮質、黒質、視床、視床下核に萎縮が認められ、歯状核への影響は軽微とのことでした。
線条体
脳には基底核と呼ばれるいくつかの核があり、主に運動調節と運動学習を担っています。基底核の入り口となる核は「線条体」と呼ばれ、脳の大脳基底核の一部で、筋肉の緊張を調節し、さまざまな細かい動きや複雑な動きを調整する役割を果たしています。線条体のγ-アミノ酪酸系中型多発性神経細胞は、大脳皮質のグルタミン酸系神経細胞と黒質のドーパミン系神経細胞の両方から投射を受けています。
中型有棘神経細胞 (MSN) は、D1型ドーパミン受容体とD2型ドーパミン受容体を発現する2つのサブタイプに分けられ、D1型は随意運動を促進する直接経路を、D2型は不随意運動を抑制する間接経路を媒介します。HDの初期には、多くのMSNがアポトーシスを起こし、D2型細胞の死滅率がD1型より大きいため、D2型MSNが減少し、不随意運動の抑制が失われ、舞踏病に発展します。HDの後期では、症状の進行に伴いD1型とD2型の両方が障害されます。この段階では、舞踏運動の症状は減少し、運動も抑制されますが、筋強直やジストニアが起こります。
研究&治療
1. 薬物療法
現在、HDの決定的な治療薬は市販されておらず、緩和的な効果を持つ薬しかありません。テトラベナジン(tetrabenazine)とデューテトラベナジン(Deutetrabenazine)は、いずれも中枢神経系の小胞モノアミン輸送体2(VMAT 2)を阻害する薬剤です。その作用機序は、VMAT 2を標的として阻害することにより、モノアミン神経伝達物質の貯蔵を遮断、抑制することです。
テトラベナジン
テトラベナジンは、半減期が短く、副作用が大きいという特徴があります。これに対し、デューテトラベナジンは半減期を大幅に延長することができ、副作用も少なく、安全性も良好です。
デューテトラベナジン
バルベナジン(valbenazine)は、成人の遅発性運動障害の治療薬として初めて(2017年に)米国FDAから承認された薬剤で、HDの舞踏病に対してより優れた抑制効果を示しています。また、選択的ドパミンD2受容体(D2R)拮抗薬であるプリドピジン(pridopidine)も、舞踏病に対する抑制効果を有しています。現在、多くの臨床試験が行われ、有望な結果が得られており、HDの新薬として注目されています。
2. 抗体医薬
VX15は、米国のバイオテクノロジー企業であるVaccinex社の製品で、ActivMAb技術プラットフォームを用いて創製された、Semaphorin 4D(SEMA4D)の生物活性を効果的に遮断できる新規モノクローナル抗体です。
SEMA4Dは、中枢神経系における自然炎症細胞(ミクログリアとアストロサイト)の両方の活性化を誘発し、HDや多発性硬化症(MS)など様々な神経炎症性/神経変性疾患の病態生理における重要な細胞制御因子であり、これらの疾患の治療のための特異的分子標的の1つです。
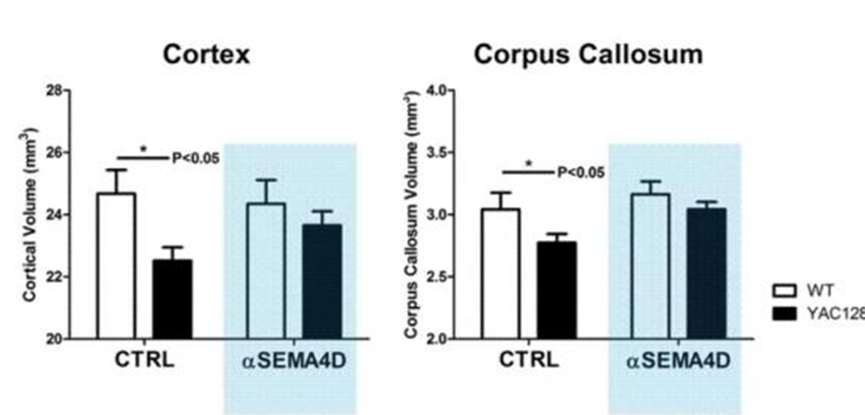
前臨床試験において、SEMA4Dを阻害することにより、脳内の神経炎を効果的に緩和し、神経細胞のアポトーシスを抑制することが実証されています。また、HDモデルマウスでは、VX15が大脳皮質および脳梁の萎縮を有意に抑制することも確認されています。
VX15は、2016年に米国FDAからオーファンドラッグとして承認されています。
3. 幹細胞治療
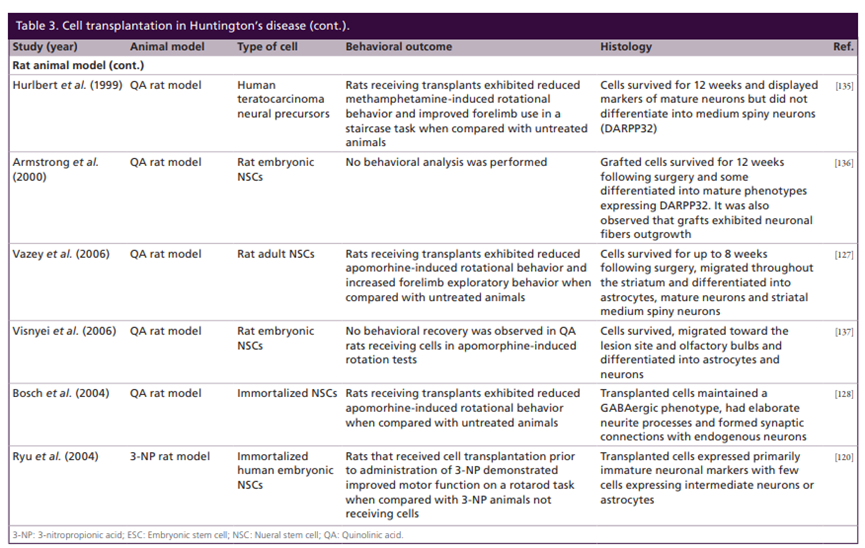
近年、幹細胞治療が、損傷した神経組織を修復し、損傷した神経細胞を置換して、症状を改善する可能性があることが科学者によって見出されています。現在、ヒト胚性幹細胞(hESC)、ヒト人工多能性幹細胞(hiPSC)、ヒト人工神経幹細胞(hiNSCs)において、一定の進展が見られています。しかし、この種の治療法にも一定の限界があり、幹細胞を体内に移植した後、その細胞の生存と分化指向性が保証されないため、さらなる研究が必要です。
このほか、遺伝子治療や先駆的な治療法もあります。次回はそれをご紹介します!ご期待ください!
参考文献:
[1] GeneticModifiers of Huntington's Disease GeM-HD Consortium. CAG Repeat NotPolyglutamine Length Determines Timing of Huntington's Disease Onset [J]. Cell, 2019, 178(4):887-900.e14.
[2] Weijie Chen, Min Ye. Advances in Clinical Research on Early Diagnosis and Treatment of Huntington's Disease [J]. Journal of Chongqing Medical University, 2019(4):3.
[3] Ross C A , Aylward E H , Wild E J , et al. Huntington disease: natural history, biomarkers and prospects fortherapeutics.[J]. Nature Reviews Neurology, 2014, 10(4).
[4] Roos R A. Huntington's disease: a clinicalreview[J]. Orphanet Journal of Rare Diseases, 2010, 5(1):40.
[5] Zuccato C, Valenza M, Cattaneo E .Molecular Mechanisms and Potential Therapeutical Targets in Huntington's Disease[J]. Physiological Reviews, 2010, 90(3):905-981.
[6] Deng Y P , Albin R L, Penney J B , et al.Differential loss of striatal projection systems in Huntington's disease: aquantitative immunohistochemical study.[J]. Journal of Chemical Neuroanatomy, 2004, 27(3):143-164.
[7] Caron NS, Dorsey E R, Hayden M R. Therapeutic approaches to Huntington disease: from the bench to the clinic [J]. Nature Reviews Drug Discovery, 2018, 17(10):729-750.
[8] Fink K D , Deng P , Torrest A , et al. Developing stem cell therapies for juvenile and adult-onset Huntington's disease[J]. Regenerative Medicine, 2015, 10(5):623-646.
