VSIG4はNLRP3に関する炎症疾患の治療の標的にされること見通し
近年の研究によるとNLRP3の炎性体の過度な活性化は様々な発病をもたらすことに関わっているがNLRP3メカニズムの表現が不十分である。中国の人民解放軍軍医大学の陳永文(チン・ヨンムン)教授と呉玉章(オ・オクチャン)氏が率いる研究チームは最近VSIG4が「マクロファージ」のNLRP3やIl-1βの表現を抑制でき、NLRP3に関連する炎症疾患の治療の標的として期待することを明らかにした。
また「VSIG4 mediates transcriptional inhibition of NLRP3 and il-1βin macrophages」という論文は今年の1月9日「Science Advances」誌に発表された。
「体外と体内の炎症が反応する間でVSIG4はマクロファージのNLRP3やIl-1βの表現を抑制できるということだ。これまでの研究に関連し、私たちはVSIG4とその関連ルートの関与性を推定し、各種の炎性疾患を治療できる可能性がある」と著者は述べた。
炎症性サイトカインのil-1βとil-18は、宿主が感染や炎症性疾患を防ぐ重要な役割を果たし、マクロファージが中心となっている。il-1β/ il18は一般的にcaspase-1のカットが必要とされ、炎性体のコントロールを活性化されるということ判明し。最も特徴的な炎性体はNLRP3でその不調による各種の変性硬化症、心血管疾患、神経変性疾患、代謝疾患などの炎症反応症候群の発生を引き起こす。そのためNLRP3の炎症体の活性化を抑制と良好バランスを保ち、宿主体内に过剰な炎症障害が発生しないようにする。
VSIG4 (v-set and immunoglobulin domain - containing 4)は免疫グロブリンの補体として、静息状态の组织がマクロファージ表面に定着し特异性表现し。補体C3の分解成分C3bとiC3bを結合することでVSIG4は病原体を撲滅する。VSIG4はT細胞の増殖を抑制することができ、Foxp3+調節可能なT細胞(Treg)の分化を促進することができる。しかし、VSIG4がNLRP3炎性成分の転写を制御できるかどうかはまだ不十分であること。
VSIG4はNLRP3やil-1βの転写を抑制する
VSIG4はNLRP3炎性体信号における潜在的な役割を研究するため、研究者はVSIG4−−/−マウスと野生型マウスから腹腔にじみ液を抽出し・マクロファージ(PEM)を分離する。qRT−PCRの研究結果ではVsig4−/−−マクロファージにはNLRP3及びil-1βのmRNAが著しく上昇した。またWestern blotの研究結果によると、Vsig4−/−−マクロファージの中でNLRP3とproIL−1βのタンパク質のレベルが向上したことも確認された。リポ多糖(LPS)は野生型と比べ、NLRP3やproil-1β蛋白の表現力を強めることができると明らかしました。(図1)。
C3bはVSIG4の天然改良体であることを考慮し、ELISAの研究結果ではLPSはマウスPEMsとRAW26 c4.7細胞C3蛋白の表現を促進することができ、研究者はこれらの条件でC3の分解成分C3bはマクロファージに含まれたVSIG4と相互作用する可能性があると推定した。この仮説を検証するため彼らはPMAで人単核細胞系thp-1を刺激し、VSIG4を誘導する。Western blotの結果によるとC3bはLPS誘導のNLRP3やproil-1βレベルの低下を刺激する。さらに検証するためにマウスVSIG4に抵抗する単一クローン抗体(MAb)のセットを開発し、VSIG4タンパク質を識別できる特定性複製(#VG11)を検証した。#VG11は、野生型大食細胞におけるLPS誘導のproIL−1βとNLRP3表現を抑制することができ、Vsig4−/−−マクロファージは一対一の抵抗処理に反応していない。これらのデータはVSIG4が抑制信号を開始し、特にマクロファージにおけるNLRP3やil-1βの表現を抑制することを意味する。
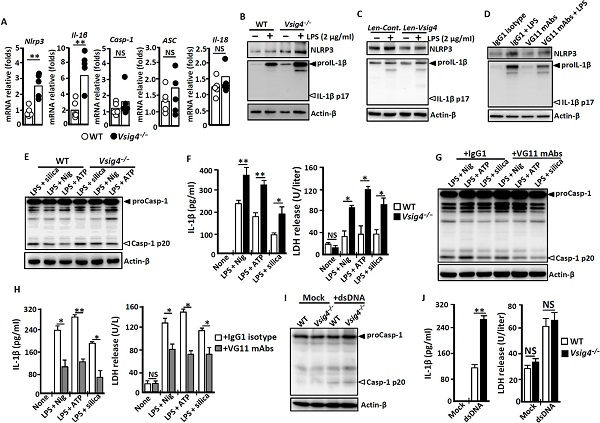
図1. VSIG4は体外でNLRP3やil-1βの転写を抑制する
VSIG4はどの信号ルートで機能するのか?
研究者はこの背景にあるシグナルルートを探索し始めた。A20はNF−米プロキシ・プロキシBの活性化を抑制しLPS誘導を低めたNlrp3プログラムの転写を受け、自然に研究対象となった。A20の沈黙がLPS誘導のNF−トロントB p65リン酸を高めたことを受け、NLRP3とil-1β蛋白が上昇を示したという。またNF−トロントB阻害剤であるBAY 11−7082はマクロファージでのLPS誘導のNLRP3とproIL−1β表現を低めた。これらのデータによるとVSIG4はA20の活性化させた送信フィードバック信号によってNF−トロントBの活性化を抑制しNLRP3とIl−1βの表現を引き下げた。
そのほかSTAT3抑制剤s3i-201はVsig4+RAW264.7細胞のLPS誘導A20を下げることできNLRP3やproil-1βの表現を向上させることができた。ほかにはJAK2阻害剤TG101348はLPS誘導のSTAT3リン酸化を低減しA20表現を抑制した。これらのデータによると、VSIG4はJAK2−STAT3−A20シグナル経路を活性化させマクロファージにNLRP3やil-1βの転写を抑制した。
次は研究者は免疫蛍光双染実験とWestern blotでVSIG4がMS4A6Dと相互作用することを分析した。免疫共殿物分析によると、MS4A6DはJAK2と直接結合し、VSIG4はVG11に一抵抗する処理はVSIG4 +RAW264.7細胞のMS4A6D/JAK2をさらに向上させ、MS4A6DはJAK2を直接活性化させることを意味する。さらにVG11は野生型マウスPEMsにおけるSTAT3リン酸化およびA20の表現を促進することができるのに対し、Ms4a6dノックアウトマウス(サイヤジェン株式会社構築)PEMは、VG11に対する反応が逆になる。一連の研究ではMS4A6DはVSIG4と相互に作用する近端類のタンパク質であり、JAK2−STAT3−A20を通して信号をさらに募集し、NLRP3及びil-1βの転写を低減することが明らかになった。
Vsig4の欠乏はマウスの病状を悪化させる?
その後研究者たちは免疫性自己免疫性脳脊髄炎(EAE)マウスをモデルにしたVsig4の欠如による影響を研究した。彼らはVsig4が野生型に比べて低レベルになると病気の発生を加速させることができることを発見した。組織病理学の分析によると、Vsig4−/−−マウスの脊髄中の浸潤性白細胞は、疾病のピーク時期に特に多かった。流式细胞术ではさらにVsig4−/−−マウスの脊髄中炎性白细胞の数が明らかに増加した。これらのデータVsig4の欠乏がEAE子鼠の病状を悪化させたことを意味する。
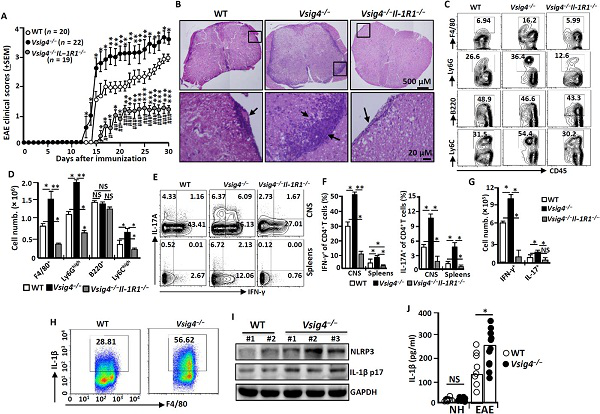
図2 Vsig4の欠乏によりマウスの病状が悪化した
但しNLRP3炎性体の過度な活性化は場合によっては保護効果を発揮するように見える。研究者らはVsig4−/−マウスを3.5%のグルカン硫酸ナトリウム(DSS)に暴露し6日間で結腸炎を誘導した。驚くことにを発見野生型マウスに比べて、DSSに露出されたVsig4−/−マウスの保存率が明らかに向上した。同時にこれらのマウスの結腸炎の重症度は野生型マウスに比べて特に低く、Vsig4−/−マウスがDSSが誘導する結腸炎に抵抗することを明らかにした。
VSIG4興奮剤はEAEの病状の進行を遅らせる
研究者はこのデータによれば、VSIG4介導抑制信号はマクロファージではNLRP3やil-1βの転写が抑制され、これはVSIG4がNlrp3に関連する炎性疾患の治療の標的になることを強調した。妥当性を検証するため彼らはEAE誘導中に野生型マウス腹腔内に激動剤VG11 mAbsまたは同タイプのIgG1 mAbsを注射した。彼らはVSIG4に対する興奮剤VG11 mAbsの刺激がNLRP3やil-1βを抑制することによってEAEの進行を遅らせることがわかった。
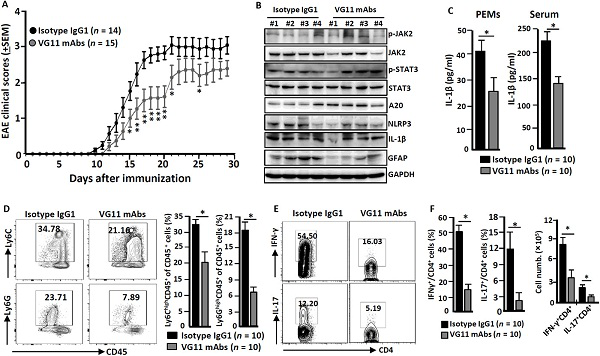
図3 VSIG4はEAEの病状の進行を遅らせる
研究者たちは野生型マウスにVsig4を強制的に表現することで、mhv-3が誘導する重症ウイルス性肝炎を改善することができることを発見した。今回の研究成果に関連し彼らはVSIG4と関連信号経路の標的は各種炎性疾患の治療に効き目があると推定した。
最新の販売促進キャンペーン:
- Cyagenノックアウトマウスライブラリ:16000種以上のKO/cKO系統マウスを所有、早くて2週間で納品
- 新型コロナウイルス(COVID-19)のワンストップ解決策:特別価格で人気標的ACE2、DPP4、TMPRSS2、CD147などのノックアウト(KO)、ノックイン(KI)及びヒト化マウスモデルを提供致します。
原文検索;
VSIG4 mediates transcriptional inhibition of Nlrp3 and Il-1β in macrophages
Science Advances 09 Jan 2019:
Vol. 5, no. 1, eaau7426
DOI: 10.1126/sciadv.aau7426
サイヤジェン株式会社について
サイヤジェン株式会社は15年間の発展を経て、全世界の数万人の科学研究者にサービスを提供しており、製品と技術は直接にCNS (Cell、Nature、Science)の定期刊を含む5,200余りの学術論文に応用されています。弊社の「ノックアウトマウスライブラリ」は低価格だけでなく、遺伝子名称を入力すれば、ワンクリックで注文まで操作できます。 ノックアウトマウス、ノックインマウス、コンディショナルノックアウトマウス、トランスジェニックマウス、GFPマウス、免疫不全マウス、無菌マウスなどのカスタマイズサービスを提供する以外、専門的な手術疾患モデルチームがあり、多種の複雑な小動物手術疾患モデルも提供できます。
